Is the European Medical Agency Experimenting on Babies with the Meningitis Vaccine Only Approved for Age 10 and Above?
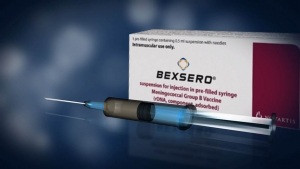
In 2016, we published an article on the dangers of the meningitis B vaccination, Bexsero, titled, Are Ineffective New Meningitis B Vaccines Causing Harm to Children? At the time of publication, according to the FDA product information leaflet, the vaccine had in fact only been approved for children over the age of ten. Despite this fact however, in the UK, the meningitis B vaccine Bexsero is being administered to infants as young as 2 months, despite the fact that we could find no evidence to support that this vaccine was safe to be administered to babies. It has been brought to our attention that there are in fact two product information leaflets on the same vaccination. However, what is different about the second product information leaflet, published on January 14, 2015, by the European Medical Agency (EMA) is that the information that it provides, is the polar opposite, of the information provided by the FDA. Is the UK government conducting clinical trials on infants, and if they are, then are parents aware of this fact?