FDA Allows Shorter Reviews for New Drug Approvals if they Promote “National Priorities” Like Operation Warp Speed Did – Iran War Updates
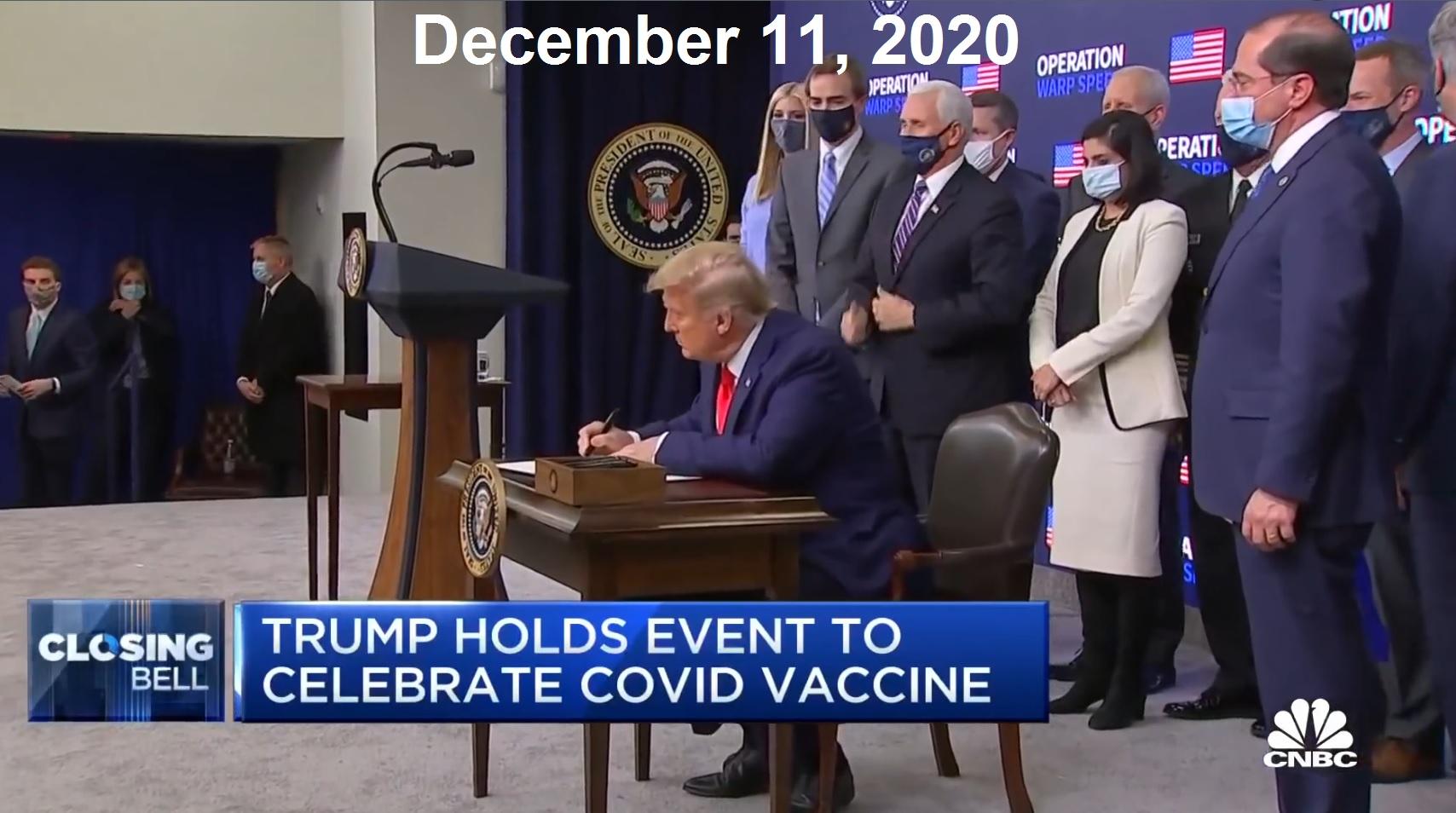
While HHS Secretary Kennedy has promised more rigorous study of drugs using higher standards and returning to the "Gold Standard of Medical Research" before they are approved, his FDA is doing the exact opposite. In a Press Release today from the FDA, Commissioner Dr. Marty Makary announced that they were now going to begin offering "vouchers" to "companies supporting U.S. national interests" for new drugs being introduced to the market, and that voucher will shorten the drug company's review time from approximately 10-12 months to only 1-2 months. In a recently published JAMA medical journal article, the FDA Commissioner said he wanted to copy the truncated process used to authorize the first COVID-19 vaccines under Operation Warp Speed. This announcement should not be too surprising, given that Mr. Kennedy stated publicly that he supported Operation Warp Speed under Trump 1.0, and would do so again. This new policy by the FDA eliminates the need to invoke a "national health crisis emergency" as Trump did with the PREP Act back in 2020. Now he can just choose which drug companies he wants to give federal funds to so they can rush their products to market, and basically use the American public as the third phase of the drug trials to see how many people these new "novel" drugs injure and kill, before making adjustments. What a financial boom to Big Pharma! Plus, updates on the war with Iran.